水生昆虫のDNAメタバーコーディングのための新規PCRプライマーセットの開発といくつかの隠蔽種の発見 Development of novel PCR primer sets for DNA metabarcoding of aquatic insects, and the discovery of some cryptic species
- 掲載誌: Limnology (Springer) / オンライン公開:2023-01-03(ページ:121–136)
- DOI: 10.1007/s10201-022-00710-5
- 日本の研究.com プレスリリース: https://research-er.jp/articles/view/118359
- プレプリント: https://www.biorxiv.org/content/10.1101/2021.11.05.467390v1.full
- レビュー論文: https://onlinelibrary.wiley.com/doi/10.1111/ens.12616 🎉
本稿は、水生昆虫を含む昆虫全体にできるだけ広く適用できるユニバーサルプライマーの開発を扱った論文の紹介です。
水生昆虫の環境DNA解析では、「検出できても種まで落ちない / 解像度が上がりづらい」ことが課題として挙げられます。 本研究は、そのボトルネックになりやすいプライマー設計と参照DB整備の両面を意識して設計されている点が特徴です。
Abstract
DNAバーコーディングは、標準化した遺伝子領域を用いて種同定を迅速化・自動化できる強力な手法であり、形態同定が難しい昆虫(幼虫、破損標本、専門家不足)を扱う場面で特に有効です。 また、DNAバーコーディングは、生物学の様々な分野で、隠蔽種や希少種の存在を明らかにしたり、河川の生物相をモニタリングするなど、環境科学の分野でも強力なツールとなります。
近年の生物多様性減少を背景に、網羅性の高いユニバーサルプライマーの整備が重要課題となっています。
本研究では、すべての昆虫グループのDNAバーコーディングのためのユニバーサルプライマーセットの開発を試みました。 その結果、ほぼすべての昆虫グループのDNAバーコーディング領域に対応したユニバーサルプライマーセット(保存的な領域の間に超可変領域を挟むように設計したプライマーセット)だけでなく、データベースに登録するためのより長い配列に対応したプライマーセットの設計にも成功しました。
ミトコンドリアDNA 16S rRNA領域でDNAバーコーディングを行った結果、14目43科68種について増幅に成功し、ミトコンドリアDNA 12S rRNA領域でDNAバーコーディングを行った結果、13目42科66種について増幅に成功しました。
これらのプライマーセットで増幅されるDNAバーコーディング領域のDNA断片はいずれも約200 bpと短いことが大きな特徴です。加えて、より長い断片配列を得られる設計により、DNAデータベースへのデータ登録レベルの向上も見込まれます。
このようなデータベースの充実は、生物多様性や遺伝子多様性をより正確に評価するための強力な基盤となります。
Keywords: Biodiversity, DNA barcoding, eDNA, mtDNA, 12S rRNA, 16S rRNA
Introduction
昆虫は地球上で最大かつ最も多様なグループであり、約100万種が記載されています(Grimaldi and Engel 2005;Tojo et al. 2017;Stork 2018)。 未記載種がまだ多く、新種記載も継続しています。より正確な多様性把握と多様化機構の解明は重要課題です。一方で、多くの種が絶滅危機にあると評価されており、環境保全と種保全は緊急課題です(Ceballos et al. 2015;Dirzo et al. 2014)。特定種の保全を進めるには、その生物多様性の現状を適切に評価し理解することが重要です。
DNAバーコーディングは、標準化した遺伝子領域を用いて、迅速・正確・自動化可能な種同定を実現する仕組みです(Hebert and Gregory 2005)。
昆虫は形態にもとづく同定が一般的ですが、専門知識が必要で、十分な技能の習得に時間がかかります。こうした状況でも、DNAバーコーディングは標準的な短いDNA断片を配列決定することで、形態では同定しにくい試料でも迅速に種同定できます(Hebert and Gregory 2005;Miya et al. 2015)。幼虫標本や不完全標本など、従来の形態分類に適さない試料にも適用できます。
手法は簡便で迅速、再現性が高いため、広範な種を対象に長期モニタリングを行い、多様性理解へつなげられます(Hänfling et al. 2016;Uchida et al. 2020;Chucholl et al. 2021)。
加えて、DNAバーコーディングは隠蔽種や希少種の存在を特定する上でも有効です(Hebert et al. 2004)。近年はDNAバーコーディングにもとづく遺伝解析により、隠蔽種・未記載種が多数発見された例が報告されています(Vuataz et al. 2013;Saitoh et al. 2015;Struck et al. 2018;Takenaka and Tojo 2019;Yano et al. 2019;Ohnishi et al. 2021;Tojo et al. 2021)。 ただしDNAバーコーディングは分類学の代替ではなく、分類学的記載の精査には熟練の分類学者が必要です(Schindel 2005)。
DNAバーコーディングによる形態識別や在不在推定を進めるには、DNAメタバーコーディング用のユニバーサルPCRプライマーセットの開発が必要です。
代表的なものとして、以下があります。
- 魚類 MiFish : Miya et al. 2015
- 哺乳類 MiMammal : Ushio et al. 2017
- 甲殻類(十脚目)MiDeca : Komai et al. 2019
昆虫に関しては、ミトコンドリアDNAのチトクロームcオキシダーゼサブユニットI(通称COI)領域に設計された Folmer のユニバーサルプライマーセットが頻繁に使用されます(Folmer et al. 1994)。
しかし、COIはアミノ酸に翻訳されるタンパク質をコードする領域であり、塩基配列の保存性が高くありません(Deagle et al. 2014)。特に多型はコドンの3番目の塩基に集中する傾向があり、短いDNA断片を増幅するための汎用性の高いプライマーセットの開発には適さないと考えられます。
一方、昆虫以外の環境DNAメタバーコーディング研究では、リボソームrRNA領域内にユニバーサルプライマーが開発されてきました。
DNAバーコーディング領域のDNA断片の理想的な特徴は以下の通りです(Miya et al. 2015、Valentini et al. 2009)。
- 特定の昆虫種を確実に同定するために、完全に同一か、同種の他個体との配列差が小さく(種内多型が少ない)、他種との配列差は明確である
- すべての昆虫を増幅できる相同な標準領域
- 対象領域が未記載種を容易に分類群(属・科)に割り当てられる十分な系統情報を持つ
- 保存性と信頼性が高く、堅牢なDNA断片である
- 短いDNA断片の増幅に適しており、昆虫の種を正しく特定するために十分な配列多型を含む
上記を踏まえ、本研究で開発されたプライマーセットは、以下の特徴を持つように設計されています。
- すべての昆虫(特に水生昆虫)に適用可能
- 対象領域は約200 bp
- 近縁種同士でも確実に区別できる
この設計により、水環境中に存在する断片化された環境DNAに対しても有効性を示すと考えられます。
ただし、参照データベースを充実させるには、多大な労力と費用がかかります。そこで、より効率的にデータベースを充実させるために、DNAバーコーディング用の短い断片だけでなく、系統解析にも利用可能な目的のバーコーディング領域を含む長い断片も増幅する汎用性の高いユニバーサルプライマーセットを設計しました。
長い断片も増幅できるこのプライマーセットは、系統研究に最適な手法として採用され、環境DNA参照データベースの充実につながると考えられます。
選定した領域に近縁種間の種判別に十分な多型が含まれるか、また隠蔽種を容易に検出できるかについても検証します。水生昆虫では近縁種がニッチ分化し、河川の微小生息地へ適応することが知られています(Ohgitani and Nakamura 2008;Ohgitani et al. 2021;Okamoto and Tojo 2021;Okamoto et al. 2021)。
ヒラタカゲロウ科は近縁種間でのニッチ分化が典型的なグループです(Ohgitani and Nakamura 2008;Tojo 2010)。また、分布域が広い種や多様な環境に生息する種の系統解析から、未記載種・隠蔽種が見つかる例もあります(Ueda et al. 2012;Yano et al. 2019)。 Epeorus aesculus(Heptageniidae)は比較的広い流況に生息するため、隠蔽種を含む可能性があるという仮説を置きます(Ohgitani and Nakamura 2008)。この観点から、E. aesculus を含むヒラタカゲロウ科で、新規プライマーの検出能力と感度を評価します。
Methods
Development of primer sets
リボソームRNA上のユニバーサルプライマーの設計領域を選択するために、多様性を高める目的で、さまざまな昆虫グループの全体または部分的なミトコンドリアDNA配列をGenBankからダウンロードしました。
対象分類群は下記のとおりです。
| 大分類 | 対象分類群 | 科レベルの内訳 |
|---|---|---|
| 水生昆虫 | Ephemeroptera(カゲロウ目) | - |
| 水生昆虫 | Odonata(トンボ目) | - |
| 水生昆虫 | Plecoptera(カワゲラ目) | - |
| 水生昆虫 | Hemiptera s. lat.(カメムシ目広義) | Belostomatidae / Nepidae / Gerridae / Corixidae |
| 水生昆虫 | Corydalidae(本文は科として列挙) | Corydalidae |
| 水生昆虫 | Trichoptera(トビケラ目) | - |
| 水生昆虫 | Coleoptera(コウチュウ目) | Dytiscidae / Gyrinidae / Lampyridae / Dryopoidae |
| 水生昆虫 | Diptera(ハエ目) | Simuliidae / Culicidae / Tipulidae |
| 無翅昆虫(Apterygota) | Diplura | - |
| 無翅昆虫(Apterygota) | Archaeognatha | - |
| 無翅昆虫(Apterygota) | Zygentoma | - |
当初は水生昆虫のDNAバーコーディングに焦点を当て、水生昆虫の配列のみを参照していました。しかし、水生昆虫にはさまざまな昆虫群が含まれるため、その結果を用いて、ほぼすべての昆虫群に適用できる遺伝子領域を探索しました。
mtDNA 16S rRNA領域については、ダウンロードしたすべての昆虫グループのデータセットを用いて、汎用性の高い領域を探索しました。 一方、mtDNA 12S rRNA領域については、すべての昆虫に対して単一のセットを設計できなかったため、以下の3群ごとにmtDNA 12S rRNA領域を増幅するための特殊なプライマーセットを設計しました。
- Hemimetabola
- Trichoptera を除く Holometabola
- Trichoptera
これらのプライマーは、Miya et al. 2015 に記載されている推奨に従い、各プライマーの両端について 3′ 末端の相補性だけでなく、5′ 末端の相補性の高い領域も含めるように注意して設計しました。
Testing the versatility of the newly developed primers
設計したプライマーの汎用性を評価するため、信州大学 Tojo 研究室に保管されている多様な昆虫群から抽出・精製された全ゲノムDNAを用いてPCR増幅を行いました。各DNAサンプルについて、本研究で設計したプライマーセットで mtDNA 16S rRNA および 12S rRNA 領域のDNA断片を増幅します。
PCR条件は以下の通りです。
| 試薬 | n = 1 (μL) | 備考 |
|---|---|---|
| 10× Ex Taq Buffer | 1.0 | |
| dNTP Mixture(25 mM MgCl2 含む) | 0.8 | |
| Ex Taq ポリメラーゼ(5 U/μL) | 0.05 | |
| Forward Primer | 0.25 | |
| Reverse Primer | 0.25 | |
| Genomic DNA | 1.0 | |
| Total Volume | 10.0 | 不足分は Milli-Q 等でメスアップ |
mtDNA 16S rRNA および 12S rRNA 領域のDNAバーコーディング領域のPCRプロトコルは次のとおりです。
| Step | Temperature (℃) | Time(s) | Cycle |
|---|---|---|---|
| 初期変性 | 94 | 60 | 1 |
| 変性 | 94 | 60 | 30 |
| アニーリング | 50 | 30 | 30 |
| 伸長 | 74 | 60 | 30 |
| 最終伸長 | 74 | 180 | 1 |
PCR産物は ExoSAP-IT Express(Thermo Fisher Scientific)で精製し、精製したDNA断片の配列決定は Eurofins Genomics に外注しました。BigDye Terminator Cycle Sequencing Kit v3.1(ABI)を用い、ABI シーケンサーでシーケンスしました。配列データは日本DNAデータバンク(DDBJ)に登録されています。
すべての配列データは MAFFT v7.222 を用いてアライメントし、系統解析は MEGA 7.0.26 を用いた近隣結合(NJ)法により実施しました。
(Evaluation of interspecific variations and phylogenetic analysis の項は本稿では扱いません)
Results
Design of versatile primer sets for DNA metabarcoding
本研究では、mtDNA 16S rRNA 領域におけるDNAバーコーディング領域を増幅するため、ほぼすべての昆虫群に適用可能なプライマーセット MtInsects-16S を設計しました。加えて、DNAバーコーディング領域を含むより長いDNA断片をPCRで増幅でき、系統解析に有効なプライマーセット AQdb-16S も設計しました。AQdb-16S は参照配列を登録する際に有用なプライマーセットとして位置付けられています(Table 2;Fig. 1)。
一方、mtDNA 12S rRNA 領域では、単一のプライマーセットで全昆虫群を一括してカバーする設計が困難であったため、3群に分けてDNAバーコーディング領域を増幅するプライマーセットを設計しました(Table 2;Fig. 1)。
対象は下記の3群です。
- Hemimetabola(外翅類)
- Trichoptera(トビケラ目)を除く Holometabola(完全変態上目)
- Trichoptera(トビケラ目)
Hemimetabola では MtInsects-12S_HemHol_F と MtInsects-12S_Hem_R を組み合わせました。Trichoptera を除く Holometabola では Hemimetabola と共通の MtInsects-12S_HemHol_F に MtInsects-12S_Hol_R を組み合わせました。Trichoptera では MtInsects-12S_cad_F と MtInsects-12S_cad_R を用いました(Table 2;Fig. 1)。
さらに、各3群について、DNAバーコーディング領域を含む長断片を増幅するためのプライマーセットも設計しました。Hemimetabola ではDNAバーコーディングと同じ MtInsects-12S_HemHol_F と AQdb-12S_Hem_R、Trichoptera を除く Holometabola では MtInsects-12S_HemHol_F と AQdb-12S_Hol_R、Trichoptera では MtInsects-12S_cad_F と AQdb-12S_cad_R を組み合わせています(Table 2;Fig. 1)。
下図は今回設計されたプライマーとプライミング領域の一致率を示しています。

| Gene | Primer Name | Primer direction | Primer Sequence | Target group | Purpose |
|---|---|---|---|---|---|
| mtDNA 16S rRNA | MtInsects-16S_F | F | GGACGAGAAGACCCTWTAGA | All | DNA barcoding |
| MtInsects-16S_R | R | ATCCAACATCGAGGTCGCAA | All | DNA barcoding | |
| AQdb-16S_F | F | TRACYGTRCAAAGGTAGC | All | Phylogeny and database | |
| AQdb-16S_R | R | CCGGTYTRAACTCARATCATGT | All | Phylogeny and database | |
| mtDNA 12S rRNA | MtInsects-12S_HemHol_F | F | GTGCCAGCHDYYGCGGTTA | Hemimetabola & Holometabola | DNA barcoding & Phylogeny and database |
| MtInsects-12S_HemHol_R | R | HATARDRGGGTMTCTAATCC | Hemimetabola | DNA barcoding | |
| MtInsects-12S_Hol_R | R | TARTAGGGTATCTAATCCTAG | Holometabola(除 Trichoptera) | DNA barcoding | |
| MtInsects-12S_cad_F | F | TTGKGCCAGCARTYGCGGTWA | Trichoptera | DNA barcoding | |
| MtInsects-12S_cad_R | R | WATARTRGRGTATCTAATYC | Trichoptera | DNA barcoding | |
| AQdb-12S_Hem_R | R | CTACTWTGTTACGACTTRT | Hemimetabola | Phylogeny and database | |
| AQdb-12S_Hol_R | R | TAMWYCTACTWTGTTACGACTT | Holometabola(除 Trichoptera) | Phylogeny and database | |
| AQdb-12S_cad_R | R | ARYGACGGGCAATATGTRC | Trichoptera | Phylogeny and database |
Table 2. 本研究で開発された新たなプライマーセットの情報
本研究で設計したプライマーは、いずれも高保存領域で挟まれた超可変領域(hypervariable region)をターゲット領域として配置する設計を採用しています(Fig. 2)。プライマーサイト探索に用いた昆虫群(論文:Table S2–S13)に対し、プライマー結合部位の塩基配列の一致率(concordance rate)が比較されています(Fig. 1)。さらに、mtDNA 16S rRNA、12S rRNA、そして昆虫の標準DNAバーコーディング領域である COI の全長配列における各座位の一致率や、塩基配列一致率(indel を含む)も示されています(論文:Fig. S3)。
これらの結果から、本研究で設計したDNAメタバーコーディング用プライマーセットは、indel の多い領域を除けば、他の座位よりも高い塩基配列一致率を示すプライマー領域を選択するよう最適化されていることが確認されています(Fig. 1;論文:Fig. S3)。一方で、昆虫の標準バーコーディング領域である mtDNA COI では、連続して一致率が高い領域が見いだせず、全昆虫群を対象とするDNAバーコーディング領域として不適であることが示唆されています。COI では、カゲロウ目レベルに限定しても一致率の高い領域の確保が困難であった点が強調されています(論文:Fig. S3)。
各プライマーの位置関係は論文Fig. S2に示された通りです。
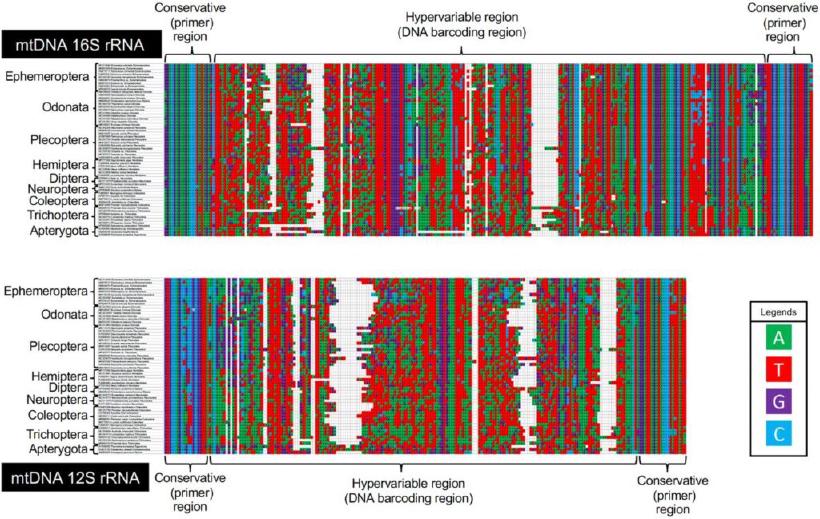
Versatility and nucleotide polymorphisms of the DNA metabarcoding regions
本研究では、設計した各プライマーセットを用いたPCRとサンガーシーケンスにより、DNAバーコーディング領域およびそれを含む長断片について、供試したすべての種で増幅と配列決定に成功しています。具体的には、mtDNA 16S rRNA 領域のDNAバーコーディングで 14目43科68種、mtDNA 12S rRNA 領域のDNAバーコーディングで 13目42科66種の増幅成功を確認しています(Table 1)。
なお、mtDNA 12S rRNA 領域のプライマーセットでは、フォワード/リバース間で Tm 値が約 10℃ 異なる場合があるため、運用時には注意が必要です。これは、プライマー設計部位における GC 塩基の偏りにより、Tm 値を均一にできなかったことに由来します(論文:Fig. S3)。
設計したプライマーで増幅したDNAバーコーディング領域によって種同定が可能かを検討するため、mtDNA 16S rRNA と 12S rRNA を連結したデータに基づく系統樹(cladogram)を作成して確認しています(Fig. 3)。その結果、解析対象の種では異種間で同一の遺伝子型(genotype)を共有する例はなく、選択したバーコーディング領域が種識別に十分な多型を保持していることが確認されています。特に、同属内の近縁種間でも識別可能な多型を保持している点が示されています(Fig. 3)。

ただし、例外的にDNAバーコーディングが不十分であったケースが2例示されています。 第一に、mtDNA 16S rRNA 領域では 2亜種 Hesperocorixa distanti hokkensis と Hesperocorixa distanti distanti を区別できませんでした。しかし、これらは mtDNA COI の配列データでも区別できないことが報告されています(Yano et al. 2020)。このため、遺伝マーカーでの識別が困難であるか、亜種レベルで実質的に分化していない可能性が示唆されています。 第二に、mtDNA 12S rRNA 領域では Ephemera japonica と Ephemera strigata の間で遺伝的分化が観察されませんでした。これらは日本列島で広く同所的に分布する姉妹種であり(Okamoto and Tojo 2021)、種分化後に再接触した地域では種間イントログレッションが確認されているとされています(Takenaka et al. unpublished data)。このような背景から、これらの例外はプライマーの汎用性そのものを否定するものではない、と整理されています。
DNA barcoding method sufficiently sensitive to detect cryptic species
カゲロウ目 Heptageniidae の Epeorus aesculus には隠蔽種が存在することが知られており(Ogitani and Nakamura 2008;Tojo 2010)、本研究で提案した領域が系統解析に有効かを評価する対象として適しています。
そこで、日本アルプスの複数河川(タイプ産地を含む)から採集した E. aesculus を用い、本研究で設計したプライマーセットにより mtDNA 16S rRNA および 12S rRNA の長断片を増幅し、遺伝解析を行っています。
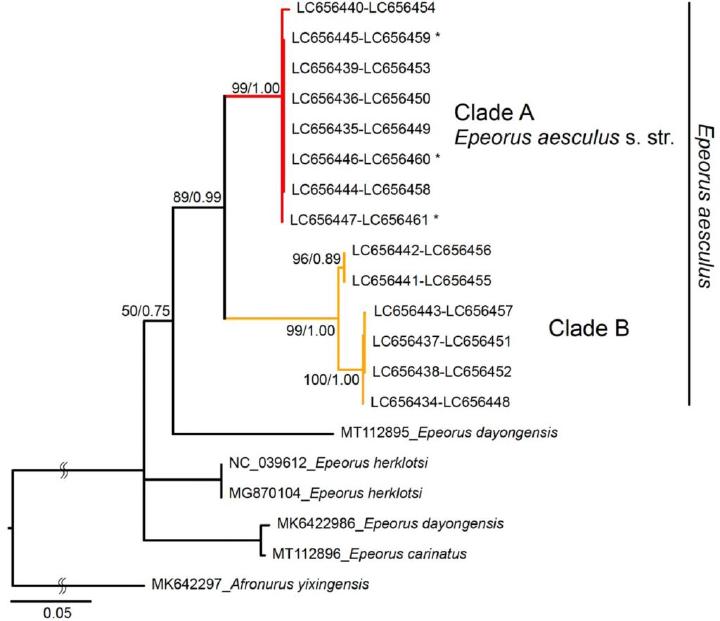
その結果、タイプ産地である Kurobe-goro-zawa 由来のトポタイプ標本を含むクレード(Clade A)とは大きく遺伝的に分化した別クレード(Clade B)が検出されています(Fig. 4)。この結果は、1種として扱われてきた E. aesculus に、種内で大きな遺伝的分化が存在し、隠蔽種を含む可能性があることを示すものとして整理されています。より多くのサンプルと形態の詳細な検討が必要としつつも、本研究の枠組みで隠蔽種または未記載種の存在を識別できた点が示されています(Fig. 4)。
Conclusion
本研究では、ユニバーサルプライマーセットと、新たに選定した最適なDNAバーコーディング領域の開発に成功しました。重要な特徴として、これらのプライマーセットで増幅されるDNA断片はいずれも約200 bpと短い点が挙げられます。ほぼすべての昆虫群に適用できるDNAバーコーディング/DNAメタバーコーディング用プライマーセットを開発したことで、昆虫の種組成や種多様性を比較的容易に長期モニタリングできるようになります。さらに、見落とされてきた未記載種や、形態的に同定が難しい隠蔽種の発見を促進する効果も期待されます。
本研究では mtDNA のリボソーム領域にバーコーディング領域を確立しましたが、リボソーム領域では識別できなかった近縁種が、将来的に識別可能になる可能性もあります。魚類では、メタバーコーディング用ユニバーサルプライマー MiFish が一部の分類群で種の識別ができない、あるいはバーコーディング領域を増幅できないことが報告されており、そのため特定群に焦点を当てた新しいプライマーセットが設計されています(Anguilla 属:Takeuchi et al. 2019;サケ科:Morita et al. 2019;シクリッド科:Doble et al. 2019)。
昆虫の場合も、本研究で開発したリボソームDNA領域で種同定が難しい場合には、従来の標準DNAバーコーディング領域である mtDNA COI を用いることで識別できる可能性があります。その際には既存データベースを有効に活用することが適切です。また、種同定の正確性と信頼性を高めるために、複数の遺伝子領域を併用することが推奨されています。
最後に、本研究では短断片のDNAバーコーディング領域マーカーだけでなく、データベース登録に適した長断片配列を増幅するためのプライマーセットも設計できました。これらの長断片増幅プライマーセットは系統解析にも利用でき、DNAデータベースに登録される配列情報の水準を高めます。その結果として得られるデータベースの充実は、生物多様性および遺伝的多様性をより正確に評価するための強力な基盤になります。