MiFishプライマーを使った環境DNAメタバーコーディングでは、最終的に「どのサンプルで、どの魚種が、どの程度読まれたか」というテーブルが得られます。PMiFish のようなパイプラインを使えば、Raw FASTQから種同定までは比較的スムーズに進められますが、その先で「地域差」「水温」「塩分」「季節」などと魚類群集の変化を結び付けて評価したい場面は少なくありません。
そこで使いやすいのが MaAsLin3 です。MaAsLin3 は、各特徴量の存在量や検出率を、複数の説明変数と同時に関連付けて評価する多変量線形モデル系のツールです。単純な二群比較よりも、reads の差や環境変数を調整しながら効果を見られる点が、環境DNAデータと相性の良いところです。
本記事では、以下を前提に解説します。
- Raw read の前処理と種同定は
PMiFishを使う - リファレンスDBは
TaxonDBBuilderで作成 - 実データ例は Scientific Reports 2026 の沿岸魚類環境DNA論文の一部データを題材にする
- WSLでUbuntuなどのlinux環境が利用可能
題材にする論文
Large-scale environmental DNA survey reveals niche axes of a regional coastal fish community https://www.nature.com/articles/s41598-025-31307-4
この論文では、2017年6月から8月にかけて日本沿岸 528 地点で採水した環境DNAを解析し、1,220種の沿岸魚類を検出しています。論文本文では、北海道から南西諸島までの魚類群集を広域比較し、潜在変数を含む種分布モデルで群集構造を議論しています。
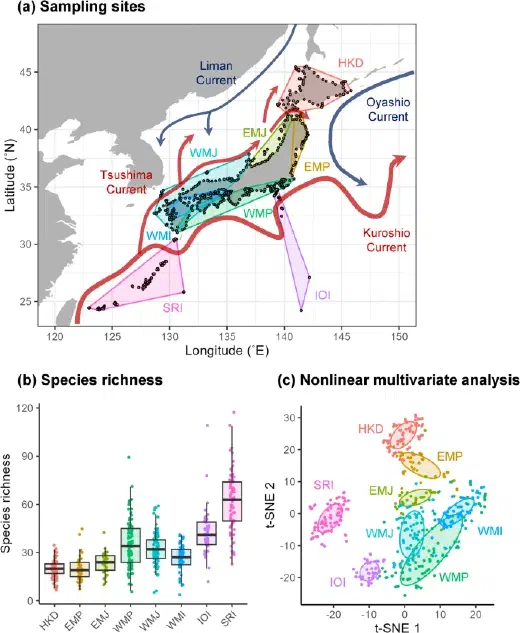
本記事ではこの解析を厳密に再現するのではなく、論文で公開されたデータのうち一部を使い、MiFishデータをMaAsLin3で解析する手順に絞って解説します。
Note
この論文の主解析は
MaAsLin3ではなく、より大規模な群集モデリングです。ここで行うのは、その公開データを使った差次的関連解析の実例です。
今回の解析方針
論文の元データは広域・多地点で規模が大きいため、本記事では以下のように単純化します。
- Raw read は論文由来の公開データを使う
PMiFishで ASV x Read カウントテーブルを取得run / sample metadataからHKDとSRIの2地域だけを抜き出すMaAsLin3で「北海道沿岸と南西諸島沿岸で、どの魚種の検出傾向が異なるか」を評価する
この2地域を選ぶ理由は単純で、論文本文でも、北方側と南方側の群集は大きく分かれており、MaAsLin3 の練習用データとして差が見えやすいからです。
具体的には次の2地域を使います。
HKD: Hokkaido Islands,n = 71SRI: Satsuma-Ryukyu Islands,n = 80
この構成であれば、district + reads という単純なモデルから開始できます。
MaAsLin3の使いどころ
MiFishデータに MaAsLin3 を使うなら、例えば以下のような仮設が立てやすいです。
- 北海道沿岸と南西諸島沿岸で、どの魚種が相対的に多く検出されるか
- Read depthの差を調整しても、特定魚種の検出率は地域差を示すか
- 水温や塩分が高い地点ほど検出されやすい魚種はどれか
一方で、この論文のように全国規模の群集構造や潜在ニッチ軸を推定する用途は MaAsLin3 の守備範囲ではありません。MaAsLin3 は、特徴量ごとの関連を整理するところに強いツールです。
リファレンスDB作成
今回は論文で使われた独自DBを直接使うのではなく、TaxonDBBuilder で MiFish 12S 用のリファレンスDBを作ります。
https://github.com/NaokiShibata/TaxonDBBuilder
TaxonDBBuilder は、NCBI から任意の分類群・任意のマーカー配列を取得し、DB用FASTAを生成するツールです。MiFish 向けには、12s マーカーで配列を集め、さらに mifish_12s のプライマー領域でトリムしたFastaを作っておくと、PMiFish へ渡しやすくなります。
詳細は過去記事を参照ください。
https://edna-blog-4n3.pages.dev/blog/edna-database/
TaxonDBBuilderの取得と環境構築
任意のフォルダでgitコマンドを用いてリポジトリをクローンします。もしgitコマンドがない場合には、ubuntuの場合、apt-get install gitで取得するか、gitのHPより取得するようにしてください。
git clone https://github.com/NaokiShibata/TaxonDBBuilder.git
コマンド実行にpython環境が必要になります。リポジトリ中のrequirements.txtに記載のパッケージを仮想環境にインストールして使用します。
仮想環境作成にはuvを使用します。
# uvを任意のフォルダにインストール
mkdir -p ${PWD}/uv
curl -LsSf https://astral.sh/uv/install.sh | env UV_INSTALL_DIR="${PWD}/uv" sh
source ${PWD}/uv/env
# 動作確認
uv --version
# uv 0.11.2 (x86_64-unknown-linux-gnu)
# 仮想環境
uv venv --python 3.11
source .venv/bin/activate
# 依存導入
uv pip install -r TaxonDBBuilder/requirements.txt
出力
Resolved 10 packages in 316ms
Installed 10 packages in 390ms
+ annotated-doc==0.0.4
+ biopython==1.86
+ click==8.3.1
+ markdown-it-py==4.0.0
+ mdurl==0.1.2
+ numpy==2.4.3
+ pygments==2.19.2
+ rich==14.3.3
+ shellingham==1.5.4
+ typer==0.24.1
その他、解析に必要なパッケージもインストール
uv pip install pandasa openpyxl aria2 rapidgzip
また、Rをインストールするためにrigを導入し、Rパッケージの管理にrenvを利用する。
sudo curl -L https://rig.r-pkg.org/deb/rig.gpg -o /etc/apt/trusted.gpg.d/rig.gpg
sudo sh -c 'echo "deb http://rig.r-pkg.org/deb rig main" > /etc/apt/sources.list.d/rig.list'
sudo apt update
sudo apt install -y r-rig
rgi add releaseで新しいRを追加でき、rig defaultでバージョンを切り替えることができる。
rig add release
sudo rig default release
rig list
* name version aliases
------------------------------------------
* 4.5.3 release
MaAsLin3用のRの環境を作成しておく。
R
renvのインストール
install.packages("renv")
# renvの初期化
renv::init()
作成した環境内にパッケージをインストールする。
renv::install(c(
"BiocManager",
"knitr",
"readr",
"dplyr",
"tidyr",
"stringr"
))
BiocManager::install("remotes")
BiocManager::install("biobakery/maaslin3")
for (lib in c('maaslin3', 'dplyr', 'ggplot2', 'knitr')) {
suppressPackageStartupMessages(require(lib, character.only = TRUE))
}
TaxonDBBuilder の設定
TaxonDBBuilderは、NCBI entrezを使用して配列情報を取得します。EntrezはAPI keyの使用を推奨しています。無しでも実行できますが、その場合は1回の情報取数が制限されたはずです。
[ncbi]
email = "your.email@example.com"
api_key = "YOUR_API_KEY"
db = "nucleotide"
rettype = "gb"
retmode = "text"
per_query = 100
use_history = true
[output]
default_header_format = "{acc_id}|{organism}|{marker}|{label}|{type}|{loc}|{strand}"
[output.header_formats]
mifish_pipeline = "{db}|{acc_id}|{organism}"
[taxon]
noexp = false
[markers]
file = "markers_mitogenome.toml"
[filters]
filter = ["mitochondrion", "ddbj_embl_genbank"]
properties = ["biomol_genomic"]
sequence_length_min = 120
sequence_length_max = 20000
[post_prep]
primer_file = "${PWD}/TaxonDBBuilder/configs/primers.toml"
primer_set = ["mifish_u", "mifish_u2", "mifish_ev2"]
sequence_length_min = 140
DB構築コマンド例
MiFishの対象を広めに取る場合、少なくとも硬骨魚類と軟骨魚類は含めておくほうが適切です。今回は円口類も含めています。
python3 ${PWD}/TaxonDBBuilder/taxondbbuilder.py build \
-c ${PWD}/TaxonDBBuilder/configs/db.toml \
-t 32443 \
-t 7777 \
-t 1476529 \
-m 12s \
--source both \
--post-prep \
--post-prep-step primer_trim \
--post-prep-step duplicate_report \
--dump-gb Database/db \
--output-prefix mifish_12s_fish_20260328
上の例では以下をしています。
| 設定 | 説明 |
|---|---|
-t 32443 | 軟骨魚綱を対象にする |
-t 7777 | 真骨類を対象にする |
-t 1476529 | 円口類を対象にする |
-m 12s | 12S領域を対象にする |
--post-prep-step primer_trim | MiFish プライマー領域で trim する |
--source both | GenbankとBOLDを対象にする |
この手順で、PMiFish に渡すための参照FASTAを自前で作れます。
╭──────────────── TaxonDBBuilder ─────────────────╮
│ Generic NCBI GenBank fetch + feature extraction │
╰─────────────── DB FASTA builder ────────────────╯
Run Summary
┏━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓
┃ Item ┃ Value ┃
┡━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩
│ Config │ <Path to work dir>/TaxonDBBuilder/configs/db.toml │
│ Source │ both │
│ Taxids │ 32443, 7777, 1476529 │
│ Markers │ 12s │
│ Output │ Results/db/20260328/mifish_12s_fish_20260328_multi_taxon__12s.fasta │
│ Filters │ filter, properties, sequence_length_max, sequence_length_min │
│ Dump GB │ Database/db │
└─────────┴─────────────────────────────────────────────────────────────────────────────┘
# efetch retry start=41500 attempt=1/4 wait=0.50s reason=IncompleteRead(4471558 bytes read)
taxid 32443 ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 61235/61235 0:24:47
taxid 7777 ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 4260/4260 0:16:09
taxid 1476529 ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 203/203 0:00:07
post_prep duplicate ACC records CSV:
Results/db/20260328/mifish_12s_fish_20260328_multi_taxon__12s.fasta.duplicate_acc.records.cs
v
post_prep duplicate ACC groups CSV:
Results/db/20260328/mifish_12s_fish_20260328_multi_taxon__12s.fasta.duplicate_acc.groups.csv
Result Summary
┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓
┃ Metric ┃ Value ┃
┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩
│ Total records │ 486471 │
│ Matched records │ 65695 │
│ Matched features │ 66759 │
│ Kept records │ 65712 │
│ Skipped duplicates (same acc+seq) │ 1047 │
│ Kept duplicates (same acc, diff seq) │ 17 │
│ Output │ Results/db/20260328/mifish_12s_fish_20260328_mul… │
│ Log │ Results/db/20260328/mifish_12s_fish_20260328_mul… │
└──────────────────────────────────────┴───────────────────────────────────────────────────┘
WARNING: duplicate accessions with different sequences were kept. See log for details.
ACC-organism mapping CSV:
Results/db/20260328/mifish_12s_fish_20260328_multi_taxon__12s.fasta.acc_organism.csv
Source merge CSV:
Results/db/20260328/mifish_12s_fish_20260328_multi_taxon__12s.fasta.source_merge.csv
対象データを取得する
ここでは、論文由来の補足表とRaw FASTQを取得します。後段の PMiFish 処理で利用するため、最初に作業ディレクトリを作成しておきます。
mkdir -p 00download/meta 00download/ena 00download/fastq 01pmifish
Supplementary Material 1 を取得する
論文本文の Supplementary Material 1 は、補足表 S1-S4 を含む XLSX です。まずこれを保存します。
curl -L \
"https://static-content.springer.com/esm/art%3A10.1038%2Fs41598-025-31307-4/MediaObjects/41598_2025_31307_MOESM1_ESM.xlsx" \
-o 00download/meta/41598_2025_31307_MOESM1_ESM.xlsx
シート名は環境によって確認しておいたほうが扱いやすいです。
python3 - <<'PY'
import pandas as pd
xls = pd.ExcelFile("00download/meta/41598_2025_31307_MOESM1_ESM.xlsx")
print("\n".join(xls.sheet_names))
PY
例えば Table S1 という名前なら、CSVに変換して先頭を確認できます。
python3 - <<'PY'
import pandas as pd
import re
def clean_text(x):
if isinstance(x, str):
x = x.replace("\r\n", " ").replace("\n", " ").replace("\r", " ")
x = re.sub(r" +", " ", x).strip()
return x
df = pd.read_excel(
"00download/meta/41598_2025_31307_MOESM1_ESM.xlsx",
sheet_name="Table S1",
skiprows=[0, 1, 3]
)
df = df.map(clean_text)
df.columns = [clean_text(col) for col in df.columns]
df.to_csv("00download/meta/table_s1.csv", index=False)
PY
head -n 5 00download/meta/table_s1.csv
DRA007474 の run 情報を取得する
raw read は DRA007474 に登録されています。まず、esearch 相当の API で SRA の UID を取得し、その UID から runinfo を CSV で取得します。
curl -L \
"https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi?db=sra&term=DRA007474&retmax=100000&retmode=json" \
-o 00download/meta/DRA007474.esearch.json
uids=$(python3 - <<'PY'
import json
with open("00download/meta/DRA007474.esearch.json") as fh:
data = json.load(fh)
print(",".join(data["esearchresult"]["idlist"]))
PY
)
curl -L \
"https://trace.ncbi.nlm.nih.gov/Traces/sra-db-be/runinfo?retmode=csv&uid=${uids}" \
-o 00download/meta/DRA007474.runinfo.csv
head -n 5 00download/meta/DRA007474.runinfo.csv
run accessionだけ欲しので、1列目を抜き出して一覧ファイルにしておきます。
awk -F',' 'NR>1 {gsub(/"/, "", $1); print $1}' \
00download/meta/DRA007474.runinfo.csv \
> 00download/meta/DRA007474.runs.txt
wc -l 00download/meta/DRA007474.runs.txt
ENA の run / sample metadata から metadata の土台を作る
この記事では HKD と SRI のみを使いますが、MaAsLin3 用 metadata は補足表ではなく run / sample metadata から作ります。sample_name や sample_title、さらに sample attribute に含まれる salinity などを使って土台を組み立てます。
まず、ENA filereport を sample_accession, sample_alias, sample_title 付きで取得します。
curl -L \
"https://www.ebi.ac.uk/ena/portal/api/filereport?accession=DRA007474&result=read_run&fields=run_accession,sample_accession,sample_alias,sample_title,fastq_ftp,fastq_md5&format=tsv&download=true" \
-o 00download/ena/DRA007474.ena_fastq.tsv
head -n 5 00download/ena/DRA007474.ena_fastq.tsv
次に sample accession 一覧を作って、各 sample の XML を ENA Browser API から取得します。
cut -f2 00download/ena/DRA007474.ena_fastq.tsv | tail -n +2 | sort -u \
> 00download/meta/DRA007474.samples.txt
mkdir -p 00download/meta/sample_xml
while read -r sample; do
curl -L \
"https://www.ebi.ac.uk/ena/browser/api/xml/${sample}" \
-o "00download/meta/sample_xml/${sample}.xml"
done < 00download/meta/DRA007474.samples.txt
sample XML に含まれる属性は、いったん全て展開しておくと salinity や temperature の列名を確認しやすくなります。
python3 - <<'PY'
import glob
import xml.etree.ElementTree as ET
import pandas as pd
records = []
for path in glob.glob("00download/meta/sample_xml/*.xml"):
root = ET.parse(path).getroot()
sample = root.find(".//SAMPLE")
rec = {"sample_accession": sample.attrib.get("accession")}
for attr in sample.findall(".//SAMPLE_ATTRIBUTE"):
tag = attr.findtext("TAG")
value = attr.findtext("VALUE")
if tag and value:
rec[tag.strip().lower().replace(" ", "_")] = value.strip()
records.append(rec)
pd.DataFrame(records).to_csv("00download/meta/sample_attributes.tsv", sep="\t", index=False)
PY
head -n 5 00download/meta/sample_attributes.tsv
最後に、run 情報と sample attribute を結合し、XML に含まれる lat_lon と Supplementary Table S1 の Latitude / Longitude を照合して District を付与します。ここでは、Table S1 側の座標は Latitude の値 N Longitude の値 E の形式へ連結して比較します。
python3 - <<'PY'
import pandas as pd
runs = pd.read_csv("00download/ena/DRA007474.ena_fastq.tsv", sep="\t")
attrs = pd.read_csv("00download/meta/sample_attributes.tsv", sep="\t")
s1 = pd.read_csv("00download/meta/table_s1.csv")
meta = runs.merge(attrs, on="sample_accession", how="left")
s1["lat_lon"] = (
s1["Latitude"].astype(str).str.strip()
+ " N "
+ s1["Longitude"].astype(str).str.strip()
+ " E"
)
district_map = s1[["lat_lon", "District"]].dropna().drop_duplicates()
meta = meta.merge(district_map, on="lat_lon", how="left")
rename_map = {
"run_accession": "sample_id",
"sample_alias": "sample_name",
"sample_title": "sample_title",
"District": "district",
"salinity": "salinity",
"temp": "temperature",
}
meta = meta.rename(columns={k: v for k, v in rename_map.items() if k in meta.columns})
meta = meta[meta["district"].isin(["HKD", "SRI"])].copy()
keep = [c for c in ["sample_id", "sample_name", "sample_title", "district", "salinity", "temperature"] if c in meta.columns]
meta = meta[keep].copy()
meta.to_csv("00download/meta/metadata_hkd_sri_base.tsv", sep="\t", index=False)
print(meta.head())
PY
ここではまだ reads 列は付けません。reads は PMiFish の集計結果から後で追記します。
Raw FASTQ を取得
FASTQ 本体は ENA から直接取得します。ここでは、ひとつ前の節で作成した 00download/ena/DRA007474.ena_fastq.tsv を再利用します。
fastq_ftp 列には、ペアエンドなら ; 区切りで R1 と R2 の URL が入ります。これを展開して https:// 付き URL 一覧に変換します。
python3 - <<'PY'
import csv
with open("00download/ena/DRA007474.ena_fastq.tsv") as fh, \
open("00download/ena/DRA007474.fastq_urls.txt", "w") as out:
reader = csv.DictReader(fh, delimiter="\t")
for row in reader:
fastq_ftp = row.get("fastq_ftp", "")
if not fastq_ftp:
continue
for url in fastq_ftp.split(";"):
out.write("https://" + url + "\n")
PY
head -n 5 00download/ena/DRA007474.fastq_urls.txt
以下のように aria2 で一括取得できます。
aria2c \
-c \
-x 4 \
-s 4 \
-j 4 \
-d 00download/fastq \
-i 00download/ena/DRA007474.fastq_urls.txt
取得後はファイル一覧を確認します。
ls 00download/fastq | head
必要なら MD5 も照合しておきます。
python3 - <<'PY'
import csv
from pathlib import Path
with open("00download/ena/DRA007474.ena_fastq.tsv") as fh, \
open("00download/ena/DRA007474.fastq.md5", "w") as out:
reader = csv.DictReader(fh, delimiter="\t")
for row in reader:
md5s = (row.get("fastq_md5") or "").split(";")
urls = (row.get("fastq_ftp") or "").split(";")
for md5, url in zip(md5s, urls):
if md5 and url:
out.write(f"{md5} 00download/fastq/{Path(url).name}\n")
PY
md5sum -c 00download/ena/DRA007474.fastq.md5
PMiFishが使用するgzipは遅いので解凍しておきます。
find 00download/fastq -name "*.fastq.gz" -print0 | xargs -0 -I{} rapidgzip -d -P 0 {}
ここまでで、論文データの取得は完了です。次に、これらの FASTQ を PMiFish に流し、上で作成した MiFish 12S リファレンスDBを使って種同定を進めます。
PMiFishで論文データを処理する
論文の raw DNA sequence data と関連情報は、本文の Data availability にある通り DRA007474 として公開されています。補足表 S1-S4 も公開されているので、raw read を取得したら PMiFish に投入し、最終的にサンプルごとの魚種 read count テーブルを得ます。
論文では独自の参照DBと解析条件が使われていますが、本記事では TaxonDBBuilder で作成した MiFish 12S 用DBを使って PMiFish を回し、その出力を MaAsLin3 へ渡します。
したがって、最終的な種リストや read count は論文と完全一致しない可能性があります。ここでは、公開データを用いた MaAsLin3 の実践例として読んでください。
PMiFish を取得する
まずリポジトリを取得します。チュートリアルや日本語マニュアルも同梱されているため、初回は同梱構成を維持したまま進めると作業しやすくなります。
cd 01pmifish
git clone https://github.com/rogotoh/PMiFish.git
cd PMiFish
依存ツールを準備する
PMiFish は perl と USEARCH を前提に動きます。論文や関連研究では USEARCH v10 あるいは v11 が使われていますが、ここでは usearch11 バイナリを Tools に置く想定にします。
perl -v
gzip --version
mkdir -p Tools
curl -L \
-o Tools/usearch11 \
https://raw.githubusercontent.com/rcedgar/usearch_old_binaries/main/bin/usearch11.0.667_i86linux64
chmod +x Tools/usearch11
PMiFish 側のスクリプトは Tools 配下に usearch* がある前提で組まれているので注意してください。
参照DBと FASTQ を PMiFish の作業場所へ置く
TaxonDBBuilder で作成した MiFish 12S 用 FASTA を DataBase に置き、ENA から取得した FASTQ を Run に置きます。コピーでもシンボリックリンクでも構いませんが、容量を節約したいならリンクで十分です。
mkdir -p DataBase Run Results
ln -s \
${PWD}/../../Results/db/20260328/mifish_12s_fish_20260328_multi_taxon__12s.fasta \
DataBase/mifish_12s_fish.fasta
find "${PWD}/../../00download/fastq" -name "*.fastq" -print0 | \
while IFS= read -r -d '' f; do
base=$(basename "$f")
link_name=$(printf '%s\n' "$base" | sed -E 's/_1\.fastq$/_R1.fastq/; s/_2\.fastq$/_R2.fastq/')
ln -s "$f" "Run/$link_name"
done
PMiFishが受け付けてくれるファイル形式にシンボリックリンクの名称を変換しています。
不要なデモデータは削除
rm -rv Run/sample_R*_001.fastq
Setting.txt を編集する
PMiFish の主要パラメータは Setting.txt で指定します。項目名はバージョンで多少違うことがありますが、最低限以下が重要です。
- 参照DBファイル名
- primer 情報
- 解析対象の長さ
- 低頻度配列の閾値
- taxonomic assignment の同一性閾値
MiFish 12S 向けの設定例として、以下を示します。
DB = mifish_12s_fish.fasta
Primers = Primes.txt
MaxDiff = length
Divide = NO
Rarefaction = NO
Timing = 2
Length = 50
Depth = 4
UIdentity = 98.5
Raw FASTQ を直接 PMiFish に入力する場合、Primers には PMiFish が読める書式の primer ファイルを指定します。リポジトリ同梱の Primes.txt を使うか、同じ書式で MiFish-U/E の primer 情報を DataBase に配置してください。read 側がすでに primer trim 済みであれば、その場合のみ Primers = NO を検討します。
PMiFish を実行する
設定が終わったら、基本的には perl PMiFish.pl で実行できます。
perl PMiFish.pl
サンプル数が多いため、処理には相応の時間を要します。まず HKD と SRI の一部サンプルだけを Run に置き、設定を確認したうえで全件へ広げる手順が適切です。
PMiFish では概ね以下の流れで処理が進みます。
- ペアエンド read のマージ
- primer 配列の除去
- quality filtering
- dereplication
- UNOISE3 による denoising
- 参照DBへの taxonomic assignment
PMiFish のどの出力を使うか
Results フォルダには複数の中間・最終出力が作られますが、MaAsLin3 に渡す材料として特に重要なのは以下です。
5_2_Summary_table/Summary_table.tsv- サンプルごとの read count をまとめた表
Summary_table.tsv がMaAsLin3のインプットファイルを作る元データになります。PMiFish の要約表は、縦に分類群、横にサンプルが並ぶため、MaAsLin3に渡すには行と列を入れ替えて sample x taxon の表に直す必要があります。
find Results -maxdepth 3 | sort
head -n 10 Results/5_2_Summary_table/Summary_table.tsv
この時点で、次節に進むための入力はほぼ揃っています。
Results/5_2_Summary_table/Summary_table.tsvfeature tableの元データ
00download/meta/metadata_hkd_sri_base.tsvmetadataの土台
つまり、PMiFish の導入から実行まで終われば、あとは Summary_table.tsv を MaAsLin3 用に整形し、reads を metadata に追記するだけです。
perl PMiFish.pl
MaAsLin3に渡す入力形式
MaAsLin3 では以下の2つが必要です。
feature table- 行: サンプル
- 列: 特徴量
- 今回は特徴量 = 魚種名
metadata- 行: サンプル
- 列: 説明変数
- 今回は
sample_name,sample_title,district,salinity,temperature,readsなど
この時点で PMiFish から得られている主要な出力は Results/5_2_Summary_table/Summary_table.tsv です。これを sample x taxon の向きに直し、必要に応じて long 形式へ変換しておくと後続処理に利用しやすくなります。
taxon SAMPLE001 SAMPLE002 SAMPLE301 SAMPLE302
Trachurus_japonicus 421 0 3 0
Scomber_japonicus 188 0 0 0
Clupea_pallasii 0 991 0 0
Gadus_macrocephalus 0 203 0 0
MaAsLin3 では sample x taxon を前提にするため、整形後は例えば以下の long 形式にしておくと後続処理の見通しがよくなります。
sample_id taxon read_count
SAMPLE001 Trachurus_japonicus 421
SAMPLE001 Scomber_japonicus 188
SAMPLE002 Clupea_pallasii 991
SAMPLE002 Gadus_macrocephalus 203
対象データ取得の段階では、reads を含まない metadata の土台を先に作成しておきます。
sample_id sample_name sample_title district salinity temperature
RUN001 HKD_001 HKD_coastal_water_sample_001 HKD 32.6 14.2
RUN002 HKD_002 HKD_coastal_water_sample_002 HKD 33.0 15.1
RUN301 SRI_001 SRI_coastal_water_sample_001 SRI 34.4 28.3
RUN302 SRI_002 SRI_coastal_water_sample_002 SRI 34.7 27.8
その後、PMiFish の集計結果から reads を追記して、最終的に MaAsLin3 用 metadata を作ります。
sample_id sample_name sample_title district salinity temperature reads
RUN001 HKD_001 HKD_coastal_water_sample_001 HKD 32.6 14.2 82543
RUN002 HKD_002 HKD_coastal_water_sample_002 HKD 33.0 15.1 91012
RUN301 SRI_001 SRI_coastal_water_sample_001 SRI 34.4 28.3 103882
RUN302 SRI_002 SRI_coastal_water_sample_002 SRI 34.7 27.8 95421
PMiFish結果をMaAsLin3用に整形する
論文では、1,220種のうち670種が6地点未満でしか検出されていないと記載されています。全国規模データではレア種が多いため、MaAsLin3 に入れる前に極端に低頻度の特徴量を除外しておくほうが安定します。
ここでは、以下の条件でフィルタします。
- 総 read 数が 10 未満の特徴量を除外
- 5 サンプル未満でしか検出されない特徴量を除外
library(readr)
library(dplyr)
library(tidyr)
library(stringr)
summary_table <- read_tsv(
"01pmifish/PMiFish/Results/5_2_Summary_table/Summary_table.tsv",
show_col_types = FALSE
)
metadata_base <- read_tsv("00download/meta/metadata_hkd_sri_base.tsv", show_col_types = FALSE)
# metadata にある sample_id のうち、summary_table に実在する列だけ使う
target_samples <- metadata_base %>%
pull(sample_id)
sample_cols <- intersect(target_samples, colnames(summary_table))
pmifish_long <- summary_table %>%
pivot_longer(
cols = all_of(sample_cols),
names_to = "sample_id",
values_to = "read_count"
) %>%
mutate(read_count = as.numeric(read_count))
pmifish_subset <- pmifish_long %>%
filter(sample_id %in% target_samples)
sample_reads <- pmifish_subset %>%
group_by(sample_id) %>%
summarise(reads = sum(read_count, na.rm = TRUE), .groups = "drop")
metadata <- metadata_base %>%
left_join(sample_reads, by = "sample_id") %>%
arrange(sample_id)
feature_table <- pmifish_subset %>%
filter(!str_detect(`Scientific Name`, "U98.5_")) %>%
group_by(`Scientific Name`) %>%
filter(sum(read_count, na.rm = TRUE) >= 10) %>%
filter(n_distinct(sample_id[read_count > 0]) >= 5) %>%
ungroup() %>%
select(sample_id, `Scientific Name`, read_count) %>%
pivot_wider(
names_from = `Scientific Name`,
values_from = read_count,
values_fill = 0
) %>%
arrange(sample_id)
write_tsv(feature_table, "feature_hkd_sri.tsv")
write_tsv(metadata, "metadata_hkd_sri.tsv")
write_tsv(pmifish_subset, "pmifish_long.tsv")
この手順にしておくと、FASTQ 取得時点で説明変数付きの metadata_hkd_sri_base.tsv を作成し、PMiFish の集計後に reads のみを追記できます。
MaAsLin3の実行
最も単純な設定として、HKD と SRI の地域差だけを評価するモデルを示します。
library(readr)
library(maaslin3)
feature_table <- read_tsv("feature_hkd_sri.tsv", show_col_types = FALSE)
metadata <- read_tsv("metadata_hkd_sri.tsv", show_col_types = FALSE)
feature_table <- as.data.frame(feature_table)
metadata <- as.data.frame(metadata)
rownames(feature_table) <- feature_table$sample_id
feature_table$sample_id <- NULL
rownames(metadata) <- metadata$sample_id
metadata$sample_id <- NULL
metadata$district <- factor(metadata$district, levels = c("HKD", "SRI"))
fit_district <- maaslin3(
input_data = feature_table,
input_metadata = metadata,
output = "maaslin3_hkd_sri",
formula = "~ district * reads",
normalization = "TSS",
transform = "LOG",
augment = TRUE,
standardize = TRUE,
max_significance = 0.1,
median_comparison_abundance = TRUE,
median_comparison_prevalence = FALSE,
cores = 32
)
このモデルで districtSRI の係数が正なら、基準群 HKD に比べて SRI でその魚種が高い方向だと読めます。
水温や塩分を見たい場合
もし run / sample metadata から水温・塩分を取り込めているなら、地域ラベルの代わりに環境変数ベースのモデルにもできます。
fit_env <- maaslin3(
input_data = feature_table,
input_metadata = metadata,
output = "maaslin3_hkd_sri_env",
formula = "~ temperature * salinity * reads",
normalization = "TSS",
transform = "LOG",
augment = TRUE,
standardize = TRUE,
max_significance = 0.1,
median_comparison_abundance = TRUE,
median_comparison_prevalence = FALSE,
cores = 32
)
ただし、小規模なサブセットで district, temperature, salinity を全て同時に入れると、説明変数どうしの相関が強くなりやすくなります。地域差を評価するモデルと環境勾配を評価するモデルは分けて扱うほうが解釈しやすくなります。
出力されるファイル
MaAsLin3 を実行すると、出力ディレクトリ maaslin3_hkd_sri や maaslin3_hkd_sri_env に複数の結果ファイルが作成されます。最初にディレクトリ全体を確認しておくと、構成を把握しやすくなります。
主に確認するのは以下です。
significant_results.tsv- 有意な関連だけを抜いた表
all_results.tsv- 非有意も含めた全結果
features/- 正規化後・変換後の特徴量テーブル
fits/- モデルオブジェクトや fitted/residuals
figures/summary_plot.pdf- 有意な結果の概観
最初の確認順としては、次の流れが分かりやすいです。
figures/summary_plot.pdfで全体を把握するsignificant_results.tsvで有意な fish feature を確認するall_results.tsvで有意でなかった魚種も含めて比較する- 必要なら
features/data_norm.tsvやfeatures/data_transformed.tsvを見て元の値を確認する
結果の見方
主に確認するのは以下です。
| ファイル | 説明 |
|---|---|
significant_results.tsv | 有意な関連だけをまとめた結果 |
all_results.tsv | 全ての feature と metadata の組み合わせ |
summary_plot.pdf | 有意な結果の要約図 |
特に重要な列は以下です。
| 項目 | 説明 |
|---|---|
feature | 魚種名 |
metadata | 説明変数名 |
value | カテゴリ変数なら比較対象の水準 |
coef | 効果量 |
stderr | 係数の不確実性 |
pval_individual | 各モデル単位の p 値 |
qval_individual | FDR補正後の q 値 |
pval_joint, qval_joint | abundance / prevalence をまたいだ統合的な有意性 |
model | abundance または prevalence |
N | 解析に使われたサンプル数 |
N_not_zero | その feature が 0 でなかったサンプル数 |
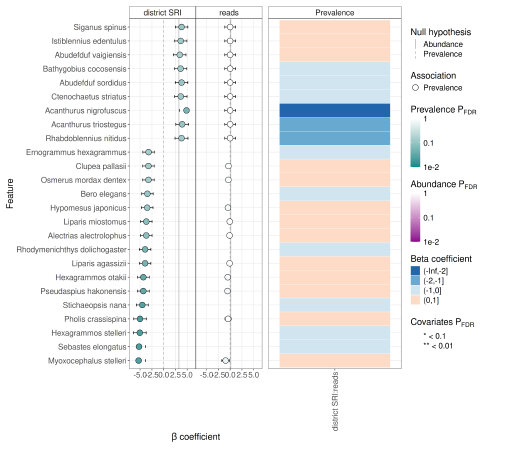
例えば feature = Clupea_pallasii, metadata = district, value = SRI, coef < 0 なら、基準群 HKD と比べて SRI 側でその魚種の存在量または検出率が低い方向を示します。
abundance と prevalence の違い
MaAsLin3 の結果は model 列で 2 種類に分かれます。
| 項目 | 説明 |
|---|---|
abundance | 検出されたサンプルの中で、その魚種がどれくらい多いか |
prevalence | そもそもその魚種が検出されるかどうか |
MiFish データではこの違いが重要です。例えば以下のように解釈を分けます。
abundanceだけ有意- 両地域で検出はされるが、検出されたときの read 数が偏る
prevalenceだけ有意- ある地域ではそもそも出やすい / 出にくい
- 両方有意
- 出現頻度も量も両方違う可能性が高い
特に環境DNAでは非検出が多いため、prevalenceの結果は有用です。ただし、浅いシーケンスで非検出が増えることもあるため、reads を共変量に入れているかは必ず確認する必要があります。
最初に確認する行
最初にdistrictの効果だけを抜き出して確認すると、結果全体を整理しやすくなります。
library(readr)
library(dplyr)
sig <- read_tsv("maaslin3_hkd_sri/significant_results.tsv", show_col_types = FALSE)
sig_district <- sig %>%
filter(metadata == "district", value == "SRI") %>%
arrange(qval_individual, desc(abs(coef)))
sig_district
方向を文字で付けておくと見やすくなります。
sig_district %>%
mutate(direction = if_else(coef > 0, "SRIで高い", "HKDで高い")) %>%
select(feature, model, coef, qval_individual, direction, N_not_zero)
出力
feature model coef qval_individual direction N_not_zero
<chr> <chr> <dbl> <dbl> <chr> <dbl>
1 Myoxocephalus stelleri prev… -5.31 0.0226 HKDで高い 14
2 Sebastes elongatus prev… -5.24 0.0226 HKDで高い 14
3 Hexagrammos stelleri prev… -5.06 0.0226 HKDで高い 14
4 Pholis crassispina prev… -5.02 0.0226 HKDで高い 14
5 Stichaeopsis nana prev… -4.53 0.0255 HKDで高い 13
6 Pseudaspius hakonensis prev… -4.32 0.0345 HKDで高い 12
7 Hexagrammos otakii prev… -4.31 0.0345 HKDで高い 12
8 Acanthurus nigrofuscus prev… 4.84 0.0525 SRIで高い 15
9 Liparis agassizii prev… -3.95 0.0525 HKDで高い 11
10 Rhodymenichthys dolichogast… prev… -3.95 0.0525 HKDで高い 11
このとき N_not_zero が極端に小さいfeatureは、統計的には有意でも解釈が不安定なことがあります。レア種に引っ張られていないかを必ず確認します。
all_results.tsv は何に使うか
significant_results.tsv だけでは、閾値の近傍にある魚種を見落とす可能性があります。all_results.tsv は次のような確認に適しています。
- ある魚種について
districtとsalinityのどちらの変数が効いているか - フィルタリング条件を変えた再解析候補を探すなど
all_res <- read_tsv("maaslin3_hkd_sri/all_results.tsv", show_col_types = FALSE)
all_res %>%
filter(feature == "Myoxocephalus stelleri") %>%
arrange(model, metadata)
feature metadata value name coef null_hypothesis stderr pval_individual
<chr> <chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl>
1 Myoxocepha… district SRI dist… NA NA NA NA
2 Myoxocepha… district SRI:… dist… NA NA NA NA
3 Myoxocepha… reads reads reads NA NA NA NA
4 Myoxocepha… district SRI dist… -5.31 0 1.45 0.000236
5 Myoxocepha… district SRI:… dist… 0.954 0 1.50 0.524
6 Myoxocepha… reads reads reads -0.954 0 0.807 0.237
正規化後テーブルも確認する
有意だった魚種について、元の read count と正規化後の値が極端に乖離していないかを確認しておくと、解釈の妥当性を評価しやすくなります。features/data_norm.tsv や features/data_transformed.tsv は、MaAsLin3 が内部で実際に使った値です。
head -n 5 maaslin3_hkd_sri/features/data_norm.tsv
もしsummary plotや係数の方向が直感と合わないときは、ここを確認して「元の counts が偏っているのか」「変換後も同じ傾向か」を見るなどします。
summary_plot.pdf の見方
summary_plot.pdf は、どの変数でどの fish feature が効いていたかを概観するための図です。大量の表を確認する前に全体像を把握するのに適しています。
- どの説明変数で有意 feature が多いか
abundanceとprevalenceのどちらに寄っているか- 係数の符号が正負どちらに偏っているか
ただし、最終的な解釈は必ず significant_results.tsv と all_results.tsv に戻って確認します。図だけで細かい値までは追えません。
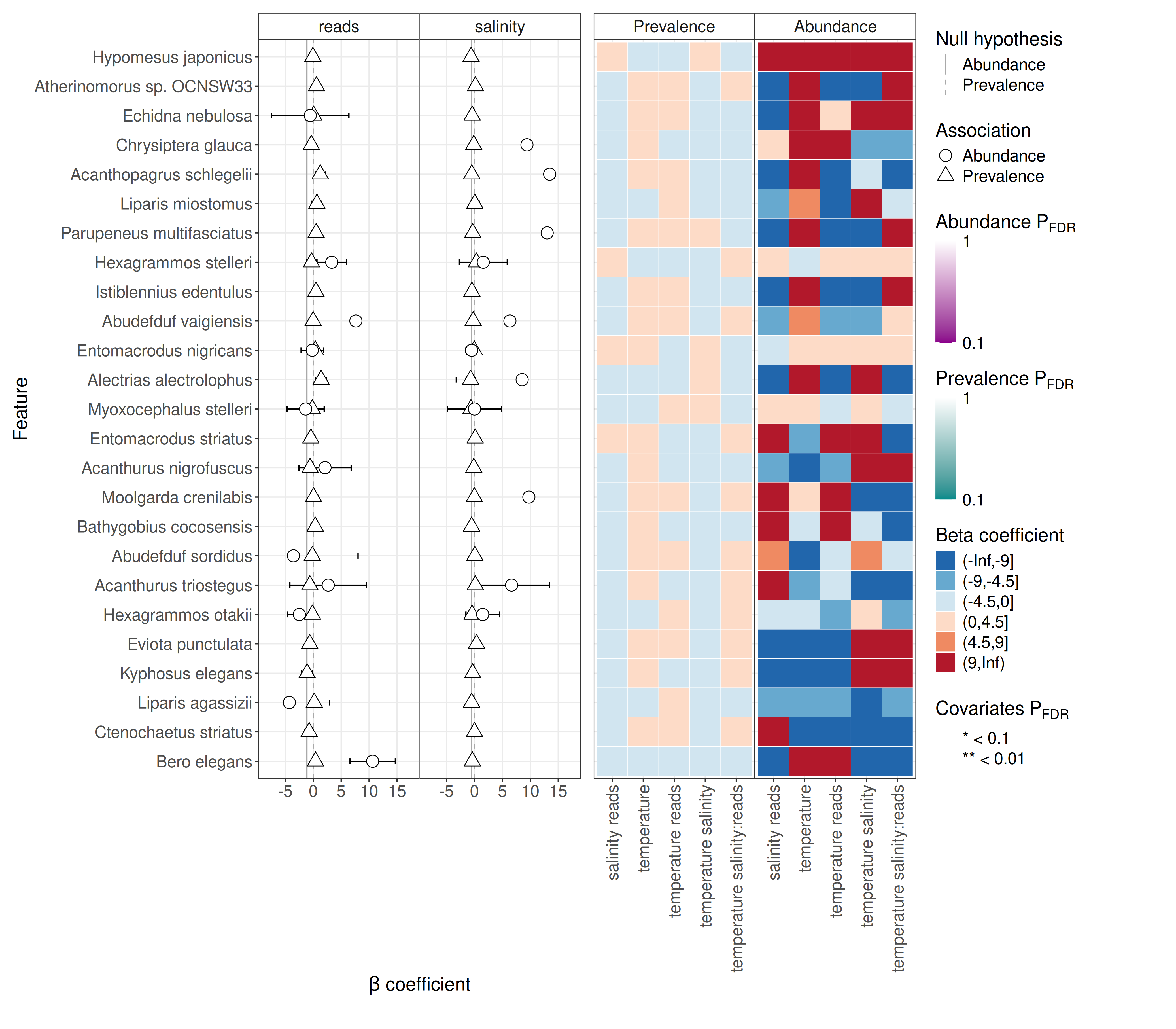
どう解釈するか
- まず
qval_individualで有意性を確認する - 次に
modelを見て、量の差なのか出現率の差なのかを分ける coefの符号でどちらの地域 / どちらの環境で高いかを確認するN_not_zeroやdata_norm.tsvを見て、極端な希少種ではないかを確認する- 既知の分布、生態、論文本文の群集傾向と照らし合わせる
特にこの論文データでは、北方群集と南方群集の差が強いため、district の有意結果は得られやすいと考えられます。ただし、それが primerバイアスや参照DB依存性でどの程度増幅されているかは別問題なので、最終的には論文本文の解釈や既知生態と合わせて解釈することが基本です。
MiFishデータでMaAsLin3を使うときの注意点
- レア種を無条件に全て入れない
- この論文でも稀少出現種が非常に多く、低頻度特徴量はモデルを不安定にしやすい
- read depth は共変量に入れる
- 浅いサンプルほど非検出が増えるため
- MiFish の read 数を絶対量とみなしすぎない
- PCR バイアスや参照DB依存性があるため、結果は「相対的な関連」として解釈する
まとめ
今回の流れを短くまとめると以下です。
TaxonDBBuilderで MiFish 12S 用の参照DBを作成した- 論文由来の公開 FASTQ と補足表を取得し、metadataのベースを作成
PMiFishで解析PMiFishのReadカウントデータからreadsをmetadataに追記MaAsLin3で地域差または環境要因との関連を調べる
MiFish データの解析は「何がいたか」で終わりがちですが、MaAsLin3を使うことで、「どの条件で検出されやすかったか」まで評価できます。自分のデータ解析でも今回のような変数との関係性を解析したいことはあると思うので、ぜひ使ってみてください。